The University at Buffalo Pseudoatom Databank
UBDB databank is derived from theoretical rather than experimental charge density distributions by fitting the pseudoatom parameters to the diffraction pattern of a large number of small molecules inserted in a pseudo-cubic elementary cell (Dominiak et al., (2007). J. Chem. Theory Comput. 3 232). The theoretical densities are obtained from G03/B3LYP/6-31G** single-point calculations on the basis of experimental geometries of small molecules taken from the Cambridge Structural Database (CSD) (Allen, (2002). Acta Cryst. B58, 380). Each atom type results from averaging over a family of chemically unique pseudoatoms, taking into account both first and second neighbors. A spawning procedure is used to ensure close transferability of aspherical atomic electron densities.
The aspherical-atom scattering factors derived from UBDB were tested in the refinement of the high-resolution (dmax ≤ 0.44 Å) and the truncated low-resolution (dmax = 0.83 Å) X-ray diffraction datasets of the tripeptide Tyr-Gly-Gly monohydrate and the hexapeptide cyclo-(D,L-Pro)2-(L-Ala)4 monohydrate (Volkov et al., (2007). Acta Cryst. D63, 160), and recently of dipeptide His-Ala monohydrate (Bąk et al., (2011). Acta Cryst. A67, 141). Application of the databank to the low resolution data, with a cut-off typical for the best available protein structures, significantly lowers the conventional R-factor, improves the bond determination and angles to within 0.002-0.003 Å and 0.09-0.17° of high-resolution values, and improves the phase angles by 2-6° compared to the standard independent atom refinement (IAM). It removes the majority of the bonding features from the residual Fourier difference maps. When freely refined, proper position of hydrogen atoms are recovered with the r.m.s. difference below 0.04 Å compared to the standard neutron X-H distances. Thermal motion parameters’ verification with the Hirshfeld rigid-bond test (Hirshfeld, (1976). Acta Cryst. A32, 239) indicates the increased physical meaning of the results.
It was already shown that the databank reproduces well electron density distribution in a number of amino acids, when compared with those calculated with conventional quantum mechanics methods at the B3LYP/6-31G** level, while requiring only a small fraction of the computational time. On the basis of the aspherical atom charge density parameters stored in UBDB accurate predictions of local and integrated properties of the electron density (Volkov, et al., (2004). J. Phys. Chem. A 108, 4283) might be drawn and so the electrostatic interaction energies. The r.m.s. deviation between the UBDB combined with the Exact Potential Multipole Method (UBDB+EPMM) and the G03/B3LYP/6-31G** results equals to 4 kJ/mol for six α-glycine dimers (Volkov, et al., (2006). J. Chem. Theory Comput. 2, 81).
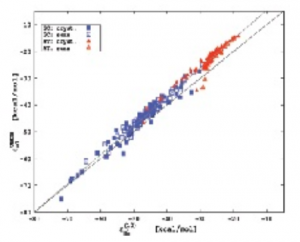
(The EPMM method (Volkov, et al., (2004). J. Phys. Chem. A 108, 4283) was developed in the course of databank creation to speed up the calculation yet still providing accurate predictions of electrostatic energies. It combines numerical evaluation of the exact Coulomb integral for short-range interactions with the Buckingham-type multipole approximation for the long-range interatomic interaction energies.) In the case of much larger molecule- antibiotic vancomycine complexes (Li, et al., (2006). Acta Cryst. D62, 639) the accuracy is slightly worse. For seven complexes of vancomycine fragment with small ligands, including dipeptides, a depsipeptide and a tripeptide, the r.m.s. deviation between the UBDB+EPMM and G03/B3LYP/6-31G** results is 31 kJ/mol. Surprisingly, the UBDB+EPMM values for vancomycin complexes are closer to these from the higher level of computation (G03/B3LYP/DZP; r.m.s.d. = 20 kJ/mol) than those from the level of theory used to generate the UBDB databank. Our recent studies on a large set of nucleic base dimers (Czyżnikowska, et al., (2010). J. Phys. Chem. B 114, 9629) have shown that the difference between values of electrostatic interaction energies computed by the UBDB+EPMM and high level (MP2/aug-cc-pVDZ) ab initio methods is 16 kJ/mol for the set comprising more than 200 Watson-Crick base pairs (Fig. 1. Left).
 |
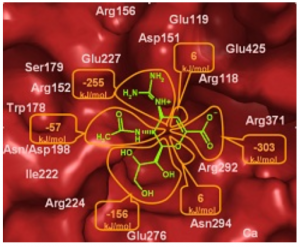 |
Fig. 1. Left, comparison of electrostatic interaction energy calculated with the UBDB+EPMM method, εelUBDB, and using the ab initio methods, εel(10), for the set of nucleic base dimers. Right, contributions to the electrostatic interaction energy of zanamivirneuraminidase complex from the six distinctive regions of the inhibitor, kJ/mol.</td |
In case of macromolecules, the UBDB+EPMM method was used to evaluate the electrostatic interaction energy of the syntenin PDZ2 domain interacting with four-residue peptides and of the PDZ2 dimer (Dominiak, et al., (2007). J. Chem. Theory Comput. 3 232), as well as the influenza neuraminidase interacting with a series of inhibitors (Dominiak et al., (2009). Acta Cryst. D65, 485). The quantitative analysis of the PDZ2 electrostatic interactions allowed to identify the most important pairwise interactions in the complexes and to explain, at least to some extent, the degeneration of ligand binding specificity. For the wide range of complexes of influenza neuraminidases, the electrostatic component of inhibition was discussed in terms of the contributions from different fragments of inhibitor molecule, (Fig. 1. Right). Also, the importance of each amino acid residue, selected structural water molecules and proximal calcium ion was analyzed. Comparative analysis of a series of wild and mutated neuraminidase complexes gave a deeper insight into the molecular basis of the influenza virus resistance to neuraminidase inhibitors caused by the Arg292Lys mutation.
The idea of building pseudoatom databank has attracted wide interest among other research groups. In the theoretical databank of Invarioms, each transferable atom is generated from charge density of a single model compound (Dittrich, et al., (2006). Acta Cryst. D62, 1325). In an experiment-based databank ELMAM, aspherical pseudoatom parameters are acquired from highly accurate experimental data for small molecules (Zarychta, et al., (2007). Acta Cryst. A63, 108; Domagała, et al., (2008). J. Appl. Cryst. 41, 1140). Very recently we compared the quality of structural and electrostatic properties obtained from the UBDB with these found for the other databases (Invariom, ELMAM and the second generation of ELMAM) (Bąk et al., (2011). Acta Cryst. A67, 141). It appeared that structural properties derived from different databanks are similar to each other. However, the values of electrostatic properties, especially electrostatic intermolecular interaction energies, differed from each other and from the reference quantum mechanics results. The UBDB bank gave the most consistent results, with r.m.s. difference in electrostatic interaction energies computed for 17 dimers equal to 17 kJ/mol and 9 kJ/mol, when compared to G03/MP2/aug-cc-pVDZ and G03/BLYP/DZP methods, respectively.